HPC for Neuroimaging
Useful Links
Introduction
This page provides the neuroimaging community with a tutorial for getting started with the HPC (High Performance Computing) cluster at the University of Arizona. The focus is on issues that are likely to be most relevant to neuroimaging. Interfaces for interacting with the HPC are much easier to use than they were years ago, and they are evolving rapidly. Look at Date Updated above to ensure that you are looking at reasonably current documentation.
The HPC is a very large cluster of linux machines at the University of Arizona. You can interact with the HPC at the command line or through a graphical user interface, but to use it effectively you should have some basic knowledge of unix.
Many big universities have similar HPCs to support research. In addition, the NSF funds other computing services for scientific research, for example Cyverse and ACCESS, both of which are nationwide. For now we’ll describe the U of A HPC.
How the HPC differs from your Desktop Computer
Unlike the computer on your desk, the HPC is a group of computers with several different host machines: bastian, filexfer, login (login node for El Gato), login2 and login3 (login nodes for Ocelote), and, of course, Ocelote, El Gato, and Puma. The storage and processing power are shared with other researchers. The different hosts provide different capabilities: For example, bastian handles security, the login nodes are for looking around and learning, filexfer handles data transfers. Ocelote, El Gato, and Puma are three different computing clusters with similar capabilities. See Compute Resources for each Cluster. All three use CentOS7 as their operating systems.
PROS
You can run a lot of image processing using your generous monthly time allocatation on the HPC.
If an image processing tool or dataset requires huge amounts of RAM, see High Memory Nodes, the HPC can provide that.
The HPC provides many high-powered NVIDIA GPUS which can really speed up our neuroimaging tools IF they are GPU-enabled. However, GPU processing is complex to set up and NOT a universally available panacea for slow processing.
The software tool has to be designed to use GPUs and that is only practical for certain types of operations.
For example, GPUs don’t help at all with Freesurfer’s recon-all but can help with FSL’s probtrackx2, Bedpostx and Eddy, all three of which provide gpu-enabled versions.
The HPC can run Apptainer containers (a.k.a. Singularity). Containers based on existing Docker containers can be created on the HPC, and several of the major BIDS containers are maintained and updated for you under
contrib/singularity/shared/neuroimaging. You do not need to use your time or space allocations to create these containers (but you can if you want to).The HPC can scale up to run lots of processes in parallel. For example, even though Freesurfer’s recon-all will still take 12-24 hours to run, you could run it on dozens of subjects at the same time.
Once you are familiar with the HPC, if it is not sufficient for your needs, you could request time on the national computing infrastructure. Again, you’ll get a lot of processing time for free.
CONS
The HPC is not HIPAA compliant for storage and processing: Data must be deidentified before you upload it to the HPC. See Deidentifying Data. HOWEVER, you can apply for access to Soteria which does provide HIPAA-compliant storage.
You have to move your data to and from the HPC for processing. See Transferring Files.
The HPC is meant for processing, not storing your data. See the Storage section below.
You do not have administrative privileges and are not allowed to run Docker
Tutorial Introduction
Below I lay out a set of steps for you to try, in what I hope is a reasonable order. Please let me (dkp @ arizona.edu) know if any of it is confusing or wrong. Don’t assume it is just you! If you get confused by the sequence of steps or the wording seems ambiguous or misleading, then other people will have the same experience and the tutorial can be improved, so I need to know. Thanks.
Sign up for an Account
Note
If you are a PI, you must follow the steps in this section to create an account and sponsor users. You do NOT have to sign on to the HPC or learn Unix or anything else, but you must create an account to get an allocation of HPC resources for your lab, and you must sponsor any staff or students who you want to have access to that allocation.
Everyone must go to the HPC Accounts Creation page for instructions on signing up.
It’ll take under an hour for your home directory to be created, but you may not have access to disk space on
/groupsfor a while longer. The HPC will send you an email when your home directory is ready.
Sign on to OOD
Once you have an account: log on to OOD (Open On Demand). Be persistent if this is your first time (it may take a few trys to be acknowledged as a bonafide user). OOD provides a dashboard interface to the HPC system:
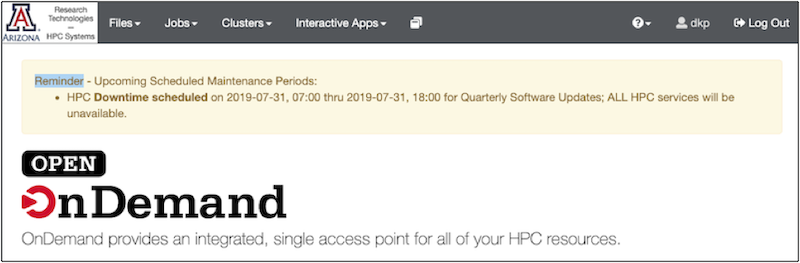
The OOD Dashboard: Along the top is a grey bar with the options: Files, Jobs, Clusters, Interactive apps.
Click each one to see what it offers:
Files offers you a graphical user interface for exploring your directories.
Jobs allows you to see the status of running batch jobs and to delete them. We’ll get to this later in the section on batch jobs.
Clusters allows you to open a command line (a.k.a shell or terminal window) on one of the machines (e.g., Ocelote, El Gato, or Puma). To use a shell, you need to know some unix commands. Note: Puma is the default shell to open. You may have trouble opening the other shells directly, but you can always open puma and switch by typing the name of one of the other machines.
Interactive Apps allows you to open a graphical desktop, Jupyter notebook or R-studio session.
We will focus on Files for now. On the top left (in the picture above), click Files. You will see options to view your home directory (at least). Select your home directory. You should see something like my home directory displayed below (but you’ll have less stuff in yours).
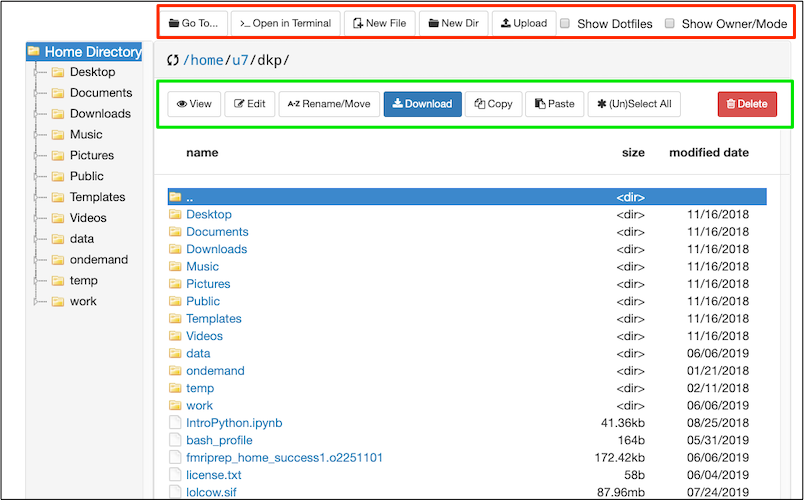
Here the OOD File Explorer is displaying the contents of my home directory in the main panel. Regardless of what is displayed in the main panel, the Home Directory is always displayed on the left of the file explorer.
In the Red rectangle (top) from left-to-right: I can use Go To.. to type in the path I want. >..Open in Terminal provides the option to open a terminal (shell) window on one of the machines. Other buttons allow me to create new files or directories, and upload or download a file (something small like a text file or image). Checking the box Show Dotfiles will reveal hidden files and folders in the current directory. These hidden files and folders are usually important for configuration. Checking the box Show Owner/Mode will display permissions. Try checking each box to see what it does.
In the Green rectangle: buttons allow various file operations, importantly the Edit button will open a text file, like a script, for editing. You can copy and paste directly from your local computer to the editor. Try various functions.
Note
You are NOT using your time allocation when you are browsing in OOD, or the File Explorer or the terminal. You won’t need your allocation time as long as you are not running anything intensive. Check out Running out of time to get a better sense of the differences between the login mode and the interactive or batch mode.
Note
Your HPC session will time out after a while (30 minutes, maybe?), so be prepared to log back in if you leave for lunch.
Warning
OOD does not get along well with the Chrome browser: If parts of the interface disappear (e.g., the editor does not work, the home directory page will not display on the sidebar, etc.), then try switching to a different browser.
The File Explorer and Editor
Try using the File Explorer and the Editor. They are really nice and easy to use.
On the top left of the File Explorer is a
Go To...button. Click on it and type/groups/dkp/neuroimaging/scripts.Select
sample_bashrc. SelectCopy.On the left, the File Explorer shows your home directory. Click
Home Directoryat the top. SelectPaste.You should now be in your home directory and have a copy of
sample_bashrcwith you.With
sample_bashrcselected, clickEdit.sample_bashrcis a bash configuration file. Parts of it will be useful to you, and other parts are configured for a different user.Let’s make a senseless edit so you can see how the editor works: Add a line like this
# helloThe
Savebutton on the upper left of the editor is activated by your change. Click it and save.There are some settings for how the editor appears on the upper right: key bindings, font size, mode, and theme.
If mode says
text, then open the dropdown menu and chooseSHindicating that this file is written in the Bash shell). This will change the text in sample_bashrc to have color coding.Try something fun like changing the color theme ; ).
Storage
Your data is not backed up. The HPC is for processing your data, not storing it.
No matter which host you are contacting, your storage is the same: Your home and groups directory are the same.
Storage in your home directory is limited: Your HPC home directory is 50 GB.
You have more space in your groups directory (500 GB per lab), and it is probably enough. If you don’t see a groups directory yet, be patient. It will be created. Later, you can look into using xdisk to sign up for as much as 20 TB of space for up to 150 days, renewable once (this allocation is at the lab level). You can no longer buy or rent additional space.
Allocation Hours
If you use interactive graphical tools (e.g. Desktop, Jupyter Notebook, RStudio), run software tools or process your data, you will need to spend your allocation hours to do so.
Your time allocation is measured in CPU hours (e.g., 1 hour of time using 1 cpu=1 CPU hour; 3 hours of time using 6 CPUs=18 CPU hours, etc). Your lab is allocated:
7000 CPU hours per month on El Gato
100,000 CPU hours per month on Ocelote, and
150,000 CPU hours per month on Puma.
This is 257,000 hours of CPU time per month, the equivalent of running 30 12-core computers 24 hours a day!
The amount of RAM you can use is limited (e.g., 4 GB per CPU on elgato, 5 GB per CPU on puma, and 6 GB per CPU on ocelote). Jobs will default to using this maximum by default.
On each machine, you can view your time allocations for that machine from a shell using va.
Jobs: Interactive and Batch
Up to this point, you have been in login mode. You can look around in the directories and create, uppload and download small files, edit a script etc. In login mode you are not using any of your monthly research allocation hours.
If you try to do a task on the login node that takes too many resources (e.g., run neuroimaging tools, build a Singularity container, archive a big directory etc.), the task will be killed before it completes.
Instead, you have to run such a task as a job. All jobs use your allocation hours and are tracked using the SLURM job submission system:
Interactive A job can be submitted to use interactive time (e.g., any of the
Interactive appslike a Desktop, Jupyter notebooks or R-studio available from OOD). You can even have interactive time at the terminal, which we explore below.Batch A job can also be submitted to run in batch mode. A batch job is one in which you submit a script to be run, and the cluster runs the job when the computing resources are available. We’ll explore this too. Batch jobs on the University of Arizona HPC use a system called SLURM.
Back on the OOD dashboard, click Jobs ➜ Active Jobs. It is probably not very interesting right now. You don’t have any jobs running on ocelote, elgato, or puma.
Check out Running out of time to get a better sense of why you need to care about creating jobs. The example focuses on building and running Apptainer/Singularity containers.
Running your First SLURM batch Job
Run a very small job so you can get a sense of how it works.
Remember you copied
sample_bashrcconfiguration file to your home area? Open it in the editor if it is not still open.About 3/4 of the way through the file, you’ll see some useful aliases, such as
interact.Open a Puma shell from the OOD dashboard:
Clusters➜Puma Shell Access. Or, in the File Explorer, select>_Open in terminal➜Puma.Copy and paste the command associated with interact:
srun --nodes=1 --ntasks=1 --time=01:00:00 --job-name=interact --account=dkp --partition=standard --pty bash -i
It is almost ready to use! You just need to change account=dkp to your account.
If you are a PI, your account will be your userid.
If you are student or staff, then your account will be your PI (sponsor) userid.
For example, if your username is
fred, but your account is sponsored bydwhite, then your account will bedwhite.If you are sponsored by several PIs, you must choose which account allocation to use.
Enter the srun command in the shell, and hit enter to run it. You should see something like this:
bash-4.2$ srun --nodes=1 --ntasks=1 --time=01:00:00 --job-name=interact --account=dkp --partition=standard --pty bash -i
srun: job 2370389 queued and waiting for resources
srun: job 2370389 has been allocated resources
(puma) bash-4.2$
You only need one node, because each node has many cpus (See Compute Resources)
The script will request a modest 1 cpu
ntasks=1It’ll run for up to one hour, unless you end it sooner. This is the
time=01:00:00.The job has a name of your choosing:
--job-name=interactThe job will use your allocation if you give it an account name and partition:
--account=dkp --partition=standard. If you do not give it a valid account name, then it cannot run on yourstandardpartition. Instead, the job will default towindfallwhich is the lowest priority job. Windfall jobs can take longer to start and can be preempted by anyone’s higher priority jobs, but they are not charged to your allocation.As soon as resources are allocated for your job, you’ll see a new command prompt
(puma) bash-4.2$.Note that it is slightly different than the original command prompt
bash-4.2$
Just because you start the interactive session with this request for 1 hour of time, does not mean you have to use the whole hour. You can delete the job. Back on the OOD dashboard, click Jobs ➜ Active Jobs. You should see your job listed:
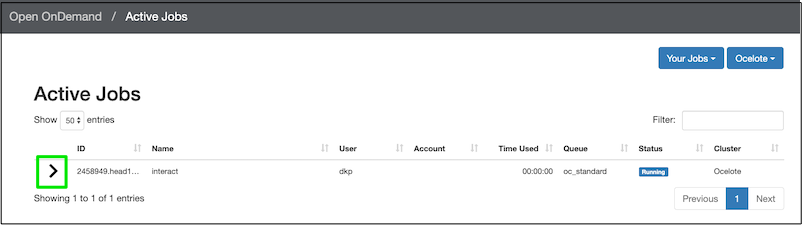
Click the disclosure triangle (in the green box) to see more details of the job. Once you have opened the job details, you’ll see a red delete button on the lower right, so you can end the job (but don’t do that just yet).
Learn about the Job
You can also learn about the job in the shell window. For example, SLURM reported my job id when the job started: 2370389. If you are not sure what your job is called, you can ask for the names of all your running jobs like this (use your own group name instead of dkp):
squeue -A dkp
Your jobs will be listed. Their status is also indicated: Q for queued; R for running.
You can also look at the resources being used by the job. To do this, you need to open another login shell and call seff with the job number:
seff 2370389
You should see output similar to this:
Job ID: 2370389
Cluster: puma
User/Group: dkp/eplante
State: RUNNING
Cores: 1
CPU Utilized: 00:00:00
CPU Efficiency: 0.00% of 00:18:32 core-walltime
Job Wall-clock time: 00:18:32
Memory Utilized: 0.00 MB (estimated maximum)
Memory Efficiency: 0.00% of 5.00 GB (5.00 GB/core)
WARNING: Efficiency statistics may be misleading for RUNNING jobs.
(puma) bash-4.2$
Cancel the Job
In addition to being able to cancel a job from the OOD dashboard, you can cancel it from the shell, like this (you have to use your job id though):
scancel 2370389
You should see output similar to this:
srun: Force Terminated job 2370389
srun: Job step aborted: Waiting up to 32 seconds for job step to finish.
(puma) bash-4.2$ Oct 23 13:42:34.987780 37489 slurmstepd 0x2b2575c51700: error: *** STEP 2370389.0 ON r1u03n2 CANCELLED AT 2021-10-23T13:42:34 ***
srun: error: r1u03n2: task 0: Killed
(puma) bash-4.2$
Job cancellation will take a moment. But then you will be charged only for the time the job was actually running.
This is a general principle, if you ask for more time and processing power than you use, you’ll only be charged for what you use. However, if you ask for a LOT of resources, it may take longer for your job to actually start.
Rerun seff to see the details of the cancelled job:
(puma) bash-4.2$ seff 2370389
Job ID: 2370389
Cluster: puma
User/Group: dkp/eplante
State: CANCELLED (exit code 0)
Cores: 1
CPU Utilized: 00:00:00
CPU Efficiency: 0.00% of 00:27:19 core-walltime
Job Wall-clock time: 00:27:19
Memory Utilized: 2.38 MB
Memory Efficiency: 0.05% of 5.00 GB
(puma) bash-4.2$
View Allocation Time on each Machine
From any shell window, type the name of the machine you want to switch to:
elgato
ocelote
puma
You can view your time allocations from the shell on each machine like this:
va
Running Lots of SLURM Jobs
You are probably not interested in the HPC for running a single small job.
for loop: Although you could write a for-loop to spawn a lot of jobs, or run job after job at the command prompt, this can overload the system scheduler and lead to problems with overall performance (see Best Practices).
BIDS allows you to process multiple subjects at a time, but the processing is sequential, and if one subject crashes, none of the following subjects will be run. In addition, you need to calculate the required CPU, memory and walltime resources for the whole set of subjects.
arrays The most efficient way to run lots of jobs is with a job array. However, it can be tricky to enter non-consecutive job names into an array.
sbatchr To address the problem of passing non-consecutive subject directory names to an array job, you can use sbatchr.
sbatchrtakes a job script and a list of subjects to run the script on.The job script only needs to allocate resources for one job, as sbatchr will create separate jobs for each subject.
The job array may run the jobs sequentially or in parallel depending on how much pressure there is on the SLURM job system from other users.
Warning
Running fMRIPrep in parallel sometimes leads to errors. Use this work around .
sbatchr and array jobs
sbatchris a wrapper script for running typical BIDS processing, it is not an official HPC script.sbatchrwill create a SLURM array job for each subject in a list of subjects (one subject per row in a text file) you pass it.Download sbatchr from bitbucket or copy it from
/groups/dkp/neuroimaging/scripts.You’ll also need
array.sh. Again, download from bitbucket array.sh or you copy it to your own area from/groups/dkp/neuroimaging/scripts.array.shis a working array script thatsbatchrcan call.array.shcreates a file with the prefixsub-for each entry in the subject list.Open your copy of
array.sh.Modify the group name
dkpto your own group name.That’s it! The script is ready to go. It’ll run the commands at the bottom under
##### BEGIN SCRIPT TO RUN ##########
Now you need a subject list:
touch my_subjects.txt
Open your empty my_subjects.txt file with the editor and add some values:
056
fred
101
mary
tom
Make sure you hit enter after the last entry to insert a blank line!
Save
my_subjects.txt. You are ready to run an array job usingsbatchrandmy_subjects.txt.It is a good idea to create a test directory where you will run this, so you can clearly identify the new files and directories that will be created.
Modify the following example to use the correct path for your copy of
array.shandsbatchr:sbatchr /groups/dkp/neuroimaging/scripts/array.sh my_subjects.txt
When it is done, you’ll see an array job file for each of your subjects:
ls -1 array*
array_slurm_my_subjects-1.out
array_slurm_my_subjects-2.out
array_slurm_my_subjects-3.out
array_slurm_my_subjects-4.out
array_slurm_my_subjects-5.out
Each array job file provides a record of what node it ran on, the date and time, and what it did:
cat array_my_subjects-1.out
/groups/dkp/test
r1u06n2.puma.hpc.arizona.edu
Sat Oct 23 15:14:43 MST 2021
JOBNAME=array_my_subjects, JOB ID: 2372273, Array Index: 1, Subject: sub-056
Detailed performance metrics for this job will be available at https://metrics.hpc.arizona.edu/#job_viewer?action=show&realm=SUPREMM&resource_id=73&local_job_id=2372273 by 8am on 2021/10/24.
There is also a log:
array_slurm_my_subjects.log
The log summarizes the job numbers for each subject:
cat array_my_subjects.log
JOBNAME=array_my_subjects, JOB ID: 2372262, Array Index: 5, Subject: sub-tom
JOBNAME=array_my_subjects, JOB ID: 2372274, Array Index: 2, Subject: sub-fred
JOBNAME=array_my_subjects, JOB ID: 2372276, Array Index: 4, Subject: sub-mary
JOBNAME=array_my_subjects, JOB ID: 2372273, Array Index: 1, Subject: sub-056
JOBNAME=array_my_subjects, JOB ID: 2372275, Array Index: 3, Subject: sub-101
(puma) -bash-4.2$
Finally, you’ll see the results of your run: files with sub- prepended to each item in my list:
ls -1 sub*
sub-056
sub-101
sub-fred
sub-mary
sub-tom
In the OOD: Active Jobs view, you can see all of the jobs have completed:
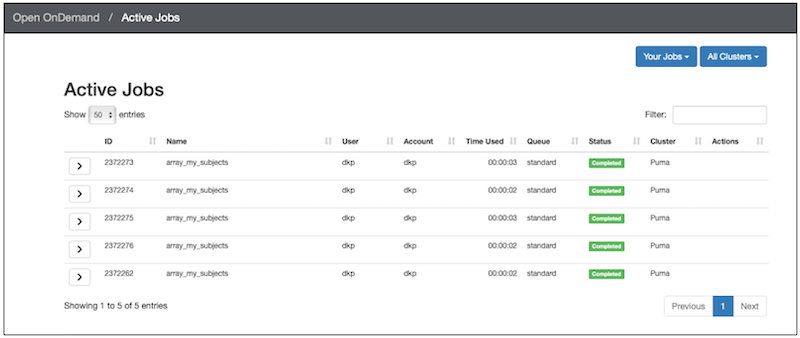
That’s it. You’ve run an array job!
Next Steps
To use your own script with sbatchr, add the following chunk of code to your script after the SLURM setup and before the code your script calls (just the way it is set up in array.sh). There is no need to change anything in this chunk of code, but I’d suggest renaming your script to indicate that it is designed for running an array job now (e.g., myscript_array.sh):
############################################################
#### BEGIN STUFF TO ADD TO ARRAY SCRIPT CALLED BY SBATCHR ####
### SLURM can specify an informative name for the output: Jobname-Jobnumber.out
#SBATCH --output=%x-%a.out
### SLURM Inherits your environment. cd $SLURM_SUBMIT_DIR not needed
### This is critical for any call to sbatchr. It gets the subject names from the list.
### Note that the subject list can be specified, but defaults to subjects.txt in the current directory if it is not specified.
### Pull filename from line number = SLURM_ARRAY_TASK_ID
Subject="$( sed "${SLURM_ARRAY_TASK_ID}q;d" "${SUBJS:-subjects.txt}")"
### The following is useful for documentation purposes.
### The array index and subject number get echoed to every output file produced by sbatch.
### Print job information to each output job
pwd; hostname; date
loginfo="JOBNAME=$SLURM_JOB_NAME, JOB ID: $SLURM_JOB_ID, Array Index: ${SLURM_ARRAY_TASK_ID}, Subject: sub-${Subject}"
### Also create a log file for the job that echos the subject number and index of each subject job to a single log file.
echo ${loginfo} >>${SLURM_JOB_NAME}.log
echo ${loginfo}
#### END STUFF TO ADD TO ARRAY SCRIPT CALLED BY SBATCHR ####
############################################################
Tip
If your script defines a subject variable, you will want to remove that definition, because the above chunk of code redefines Subject.
Having defined the variable Subject in the above chunk of code, Subject can then passed to the subsequent bash commands, e.g.:
echo ${Subject}
In summary, sbatchr creates a SLURM array job for each subject. Because each subject is a separate job, all you need to know is what cpu and time resources one subject job requires. Whenever there is room in the HPC job queue to run one or more of your subject jobs, those jobs will start.
Transferring Files
You can use the command line or graphical tools to transfer data to and from the HPC. Your allocation time is NOT used for transferring files, however, if you try to transfer large files or lots of files on the login node, your transfer will be killed. Options are described in detail on the Transferring Files page.
Tiny Files
Small files can be moved using Upload and Download in the OOD file explorer. For example, this should work for uploading or downloading a text file or a single anatomical image. If you ask to transfer something that is too big, it’ll just fail (but may not give a good message), so be suspicious and check.
Medium Sized Files
Here’s an example of using scp from the commandline to transfer a large Apptainer/Singularity container from my local mac to the HPC. It is also be a reasonable solution for a single subject BIDS dataset:
scp -v bids_data.zip dkp@filexfer.hpc.arizona.edu:/groups/dkp/shared
I have to indicate who I am on the hpc: dkp. The transfer is handled by filexfer.hpc.arizona.edu. But never fear, the file will end up in my /groups/dkp/shared directory as I specify. I have 500 GB of space in /groups/dkp and only 50 GB in my home directory, so I generally will NOT want to transfer imaging files to my home directory. This is a reasonable solution for data transfers of less than 100 GB, though not as fast as Globus.
Big Files and Directories: Globus
Globus is the preferred tool for transferring data to and from the HPC, and even between directories on the HPC.
There is a description of the Globus setup on the Transferring Files page.
More information about using Globus to transfer files can be found on the Globus Getting Started page.
Globus provides a downloadable program Globus Personal Connect for your local computer so you can treat your computer as an endpoint. You interact with the endpoints in a browser window with two panels (e.g., one panel for your local machine and one for the HPC).
Warning
Check the preferences in your locally installed Globus Connect Personal to ensure that you have access to the directories you need (by default, you only have access to your home directory). In addition, you must explicitly tell Globus (Under its Preferences) to allow you to write to these directories.
Warning
Globus does not transfer symbolic links. That is, if ALink points to AFile, then Afile will be transferred, but Alink will not. If you need symbolic links, you’ll have to tar up your directory locally and then untar it on the HPC. It turns out that tarring (archiving) your directories before transfer is a good idea anyhow.
Tip
If you have lots of files and directories (e.g. BedpostX directories), Globus slows to a crawl. For example, a Globus transfer of my ~100 GBs of data: (150 subjects in a directory containing BedpostX results) was less than ½ way done a day later. Unfortunately, zipping or tarring the datasets take some time and you’ll have to run such archiving procedures as batch jobs. Here is my experience: Tar alone for the 100 GB directory (no zipping) took 7 minutes and resulted in a 92.75 GB file: tar -cvf fsl_dwi_proc.tar fsl_dwi_proc. Tar with zip (z) took 1 hour and 16 minutes and resulted in a 90.99 GB file: tar -cvzf fsl_dwi_proc.tar fsl_dwi_proc Thus tar+zip saved me only ~2% on file size, but took almost 11 times longer to archive. Zip by itself took a long time as well. Given that the NIfTI images are all gzipped anyhow, adding z to the tar command just adds time and complexity to the command without much useful effect. Using tar without the z is the way to go for our NIfTI data. After the data is zipped, the file took only minutes to transfer. Finally, use an interactive session for tarring or risk corrupting and losing everything!!
Deidentifying Data
Until the HPC storage is HIPAA compliant, your data must be deidentified before being uploaded. After converting it to BIDS, use BIDSonym to deidentify your data. You can download a BIDSonym Google Cloud shell lesson.
Neuroimaging Software on the HPC
The following tools are available to everyone.
Apptainer (Singularity)
- HPC Administrators have provided the neuroimaging community with a dedicated area to store our BIDS Apptainer/Singularity containers: /contrib/singularity/shared/neuroimaging.
Available containers includes fMRIPrep, MRIqc, and QSIprep (You must be in interactive mode to view or access this resource).
Example scripts Examples of running the BIDS containers are in /groups/dkp/neuroimaging/scripts. These should be copied to your own bin directory and modified to suit your needs.
Learn more about the available Apptainer/Singularity Containers
Matlab Tools
Several Matlab Neuroimaging toolboxes are maintained in /groups/dkp/neuroimaging/matlab, including SPM12, GIFT, CONN, and supporting toolboxes.
Starting Matlab on HPC:
Interactive Matlab App Start Matlab directly from the OOD interactive apps page.
Start Matlab from the Interactive Desktop If you want tools other than Matlab in one session, it might be better to start the interactive desktop from OOD. After your virtual desktop starts, open a terminal (there’s an icon on the upper left, or you can right-click the desktop and choose terminal). You must load the matlab module
module load matlab
After loading the matlab module, you can access the Matlab commandline with the graphical interface:
matlabOr without without the Graphical interface:
matlab -nojvm -nosplash
Note that by default Matlab reads the path definition pathdef.m in the present working directory. It can be helpful to start matlab in the location where your pathdef.m is already set up. Matlab always adds ~/Documents/MATLAB to your path if you want to use that.
Matlab Path: The typical method of setting the matlab path through the matlab graphical interface does not persist between sessions. An alternative is to add the MATLABPATH to the .bashrc as shown in the configuration below.
Configuration
Add the following to your .bashrc to facilitate accessing FSL and the tools in the bin:
# Add an environment variable for the directory containing the apptainer/singularity containers
export SIF=/contrib/singularity/shared/neuroimaging
# Add an environment variable to the neuro directory
export NEURO=/groups/dkp/neuroimaging
# Add any number of directories to the Matlab path. This persists between sessions.
# In this example, I add spm12 and CONN. Note the standard colon for separating paths.
export MATLABPATH=/${NEURO}/matlab/spm12:${NEURO}/matlab/conn
# Add an environment variable for FSL
FSLDIR=${NEURO}/fsl
# Add an alias to invoke DiPy
alias dipy_venv='module load python/3.11/3.11.4; source /groups/dkp/neuroimaging/dipy/bin/activate; export MESA_GL_VERSION_OVERRIDE=3.3'
# caution: the MESA_GL_VERSION_OVERRIDE can cause havoc for matlab graphics!
# IF you want to plot in Matlab, you need to unset this variable first.
# Matlab version 2023b and later are not yet tested
# Add an environment variable to point to selected DiPy data (useful for testing)
export dipy_data='/groups/dkp/neuroimaging/datasets/dipy_data'
# Add tools to the path
PATH=$PATH:${NEURO}/bin:${NEURO}/bin/Mango:${NEURO}/bin/workbench/bin_rh_linux64:${FSLDIR}/bin:${NEURO}/bin/dsi-studio
# source the FSL configuration
source ${FSLDIR}/etc/fslconf/fsl.sh
export FSLDIR PATH
# run mrview from the mrtrix3 container
alias mrview='apptainer run ${SIF}/MRtrix3.sif mrview'
# run surfice from the surfice container
alias surfice='apptainer run ${SIF}/surfice.sif'
# Use this function to call mrtrix with an argument
# specifying one of the command line tools to run, e.g., mrtrix3 mrcat
mrtrix3 () { apptainer run ${SIF}/MRtrix3.sif $* ; }
If you have this in your .bash_profile, then you ensure that your .bashrc will get sourced
# .bash_profile
# Get the aliases and functions
if [ -f ~/.bashrc ]; then
. ~/.bashrc
fi
DataLad
DataLad is also available as a module from an interactive desktop Start a terminal window on the desktop:
module load contrib
module load python-virtualenvs/datalad
Datalad is now available by typing datalad
DiPy
DiPy tools, including the Horizon viewer, can be tested as follows:: Start a terminal window on the desktop:
# check dipy install
pip show dipy
# check dipy install
pip show dipy
# check fury install (fury underlies the Horizon viewer)
pip show fury
# get help
dipy_info -h
# View a NIfTI image in Horizon
dipy_horizon /groups/dkp/neuroimaging/datasets/dipy_data/mni_template/mni_icbm152_t1_tal_nlin_asym_09a.nii
# View streamline in Horizon: (--cluster gives you extra tools)
dipy_horizon --cluster /groups/dkp/neuroimaging/datasets/dipy_data/bundle_atlas_hcp842/Atlas_80_Bundles/bundles/VOF_L.trk
# exit the virtual environment
deactivate
Consult the online DiPy documentation for more information
Freesurfer
Like Matlab, FreeSurfer is available as a module from an interactive desktop Start a terminal window on the desktop:
module load Freesurfer
module list
Thanks to Adam Raikes and Dima for setting up Freesurfer!
Optional Section: SSH TO HPC
Feel free to skip this section if you are not especially interested right now. If you prefer the command line, you can ssh to the HPC e.g.,:
ssh dkp@hpc.arizona.edu
Of course, you’ll need to put in your own username, which probably is not dkp, And then you’ll go through the authentication process:
Password:
Duo two-factor login for dkp
Enter a passcode or select one of the following options:
Duo Push to XXX-XXX-XXXX
Phone call to XXX-XXX-XXXX
SMS passcodes to XXX-XXX-XXXX (next code starts with: 2)
Passcode or option (1-3): 1
Success. Logging you in...
Last login: Sun Jan 21 19:16:14 2018 from http://66-208-204-2-pima.hfc.comcastbusiness.net
***
The authenticity of host 'hpc.arizona.edu (128.196.131.12)' can't be established.
ECDSA key fingerprint is SHA256:ujL8gL1Y7A8hvKmQWbNNFzBoukcOtmYliHuIuGhCsVA.
Are you sure you want to continue connecting (yes/no/[fingerprint])? yes
Warning: Permanently added 'hpc.arizona.edu,128.196.131.12' (ECDSA) to the list of known hosts.
Last login: Thu Dec 29 17:48:41 2022 from on-campus-10-138-74-223.vpn.arizona.edu
This is a bastion host used to access the rest of the RT/HPC environment.
Type "shell" to access
This gets you to gatekeeper. Type shell (like it says) to get to the normal shell prompt.
Optional Section: Return to a Previous Terminal Session Using Screen
On your personal mac or linux computer, each terminal is open in a particular directory. You can scroll back and see the commands you ran, and you can watch the output of a long process as it appears. If you log in to the HPC, you have this up until your terminal session ends (either you close it or it times out). When you log back in later, you are at a new fresh terminal that has lost this potentially valuable information. Fortunately, there is a unix commandline tool called screen that can help with this. Screen saves sessions so you can reconnect later, even though you may have closed your HPC session. Your process keeps running in the screen and you can check on how it is getting along by attaching to it. Here is a brief tutorial on getting started with screen.
Let’s see if you have any screens (probably not):
screen -list
No Sockets found in /var/run/screen/S-dkp.
Assuming you are NOT currently in a screen session (and you aren’t if you’ve never done this before), you can create a new screen session like this:
screen
You will be switched to that new screen. You can display the name of your current working screen session like this:
echo $STY
1851.pts-4.login2
The screen has a 3-part name: screenprocessID.sessionname.host e.g., 27589.pts-4.login2
After creating the session, you should be able to refer to it with its screenprocessID or sessionname.hostname or both: (1851.pts-4.login2 or 1851 or pts-4.login2).
Detach from a screen Let’s say you don’t want to be in the screen anymore, but you want to be able to return to it later. You should detach from the screen:
screen -d 1851
Note
If you have only one attached screen, then you can simply type screen -d, but as soon as you have multiple attached screens, you need to be specific.
Now look at the state of things:
screen -list
There is a screen on:
1851.pts-4.login2 (Detached)
1 Socket in /var/run/screen/S-dkp.
echo $STY
Your screen exists, but is detached. echo $STY returns nothing because you are no longer in a screen session.
You can also create a screen with a meaningful sessionname, and you will be switched into it. In this case, the name is 2 parts: screenprocessID.sessionname:
screen -S fred
echo $STY
4032.fred
You can list your screens from inside a screen session. If we list the screens from inside fred, we see that fred is attached:
screen -list
There are screens on:
1851.pts-4.login2 (Detached)
4032.fred (Attached)
The fred screen can be referred to as 4032.fred or 4032 or fred. Let’s detach from fred, and then check that we are not in a screen session with echo $STY:
screen -d fred
echo $STY
There are screens on:
1851.pts-4.login2 (Detached)
4032.fred (Detached)
Both screens are now detached.
Re-attach to a screen session:
screen -r fred
echo $STY
4032.fred
From fred, create a third screen, joe. We should be switched to joe and both joe and fred should be attached. Check with echo $STY and list the screens:
screen -S joe
echo $STY
screen -list
There are screens on:
27376.joe (Attached)
1851.pts-4.login2 (Detached)
4032.fred (Attached)
3 Sockets in /var/run/screen/S-dkp.
Use -x instead of -r to switch to the attached screen:
screen -x joe
Warning
If you create screens within other screens (as we created joe from within fred) you can get into trouble. Ctrl AD can get you out, as you’ll probably do this at some point. However, it is best to detach from one screen before creating a new one.
When you have lost your prompt! From within a screen session that is busy or messed up, you can press Ctrl AD (Hold control, and press A and then D; these are not uppercase).
Once you have started a long process, like running a neuroimaging container, you can use Ctrl AD to detach from the screen (or you can just close the HPC browser tab and your screens will be preserved).
Kill Perhaps you have created a lot of screens. You’d like to get rid of these screen sessions altogether (not just detach from them). Here’s my current list:
screen -list
here are screens on:
1851.pts-4.login2 (Detached)
4032.fred (Attached)
27376.joe (Attached)
3 Sockets in /var/run/screen/S-dkp.
Note
Your situation may be different depending on how much you’ve mucked about creating and detaching. But you probably need some cleanup.
Here we kill a screen and then list the screens to ensure that it is gone:
screen -X -S 1851 kill
[screen is terminating]
screen -list
There is a screen on:
4032.fred (Attached)
27376.joe (Attached)
2 Socket in /var/run/screen/S-dkp.
Note that 1851 is no longer listed after being killed. Both detached and attached screens can be killed. In additon, when you are in a screen session, you can exit to both detach and kill the session:
screen -r fred
exit
[screen is terminating]
screen -list
There is a screen on:
27376.joe (Attached)
1 Socket in /var/run/screen/S-dkp.
Note
When you terminate a screen, the prompt may jump to the bottom of the terminal window!
There is more, but this should get you started. BTW, the screen utility may be lurking on other machines you use, like your mac.