HeuDiConv
Introduction
HeuDiConv (Heuristic Dicom Conversion) provides sophisticated and flexible creation of BIDS datasets. It calls dcm2niix to convert the DICOMS to NIfTI with sidecar JSON files. It also produces the additional files that BIDS expects (e.g., CHANGES, dataset_description.json, participants.tsv, README etc.). Similar tools include:
See also this excellent example of working through the whole process.
HeudiDiConv runs in a container (Docker or Singularity):
On this page, I provide tutorials for two of the heuristic files provided with HeuDiConv.
The first heuristic is called heuristic.py.
The second is called reproin.py.
heuristic.py is better if you already have DICOM files to convert. However, heuristic.py requires that you modify a Python module to specify your particular naming scheme to HeuDiConv. This process is detailed below with explicit examples for a typical U of A sequence. It isn’t horrible. Don’t assume that the Region and Exam assigned at the scanner will be available to heuristic.py in the same way they are available to reproin.py.
reproin.py takes a different approach: it requires you to follow a particular naming scheme at the scanner instead of modifying a Python module. If you have not yet named your scanner sequences, then it is worth using the reproin naming scheme. Overall, reproin is easier IF you have the opportunity to name the DICOM sequences at the scanner. Even if reproin does not work, it is always useful to name the DICOMS at the scanner in a way that helps you think about exporting to BIDS.
reproin.py is “part of the ReproNim Center suite of tools and frameworks. Its goal is to provide a turnkey flexible setup for automatic generation of shareable, version-controlled BIDS datasets from MR scanners.”
Links
A number of useful resources are available for you to consult. Here are the ones I’ve found.
Stanford Center for Reproducible Neuroscience Tutorials.
heuristic.py
reproin.py
Datasets
A test dataset to use with the heuristic.py tutorial on this page sub-219_dicom.zip
A test phantom dataset to use with the reproin tutorial on this page reproin_dicom.zip
Lesson 1: Running heuristic.py
Download and unzip sub-219_dicom.zip. You will see a directory called MRIS.
Under the MRIS directory, is the dicom subdirectory: Under the subject number 219 the session itbs is nested. Each dicom sequence folder is nested under the session. You can delete sequences folders if they are of no interest:
dicom └── 219 └── itbs ├── Bzero_verify_PA_17 ├── DTI_30_DIRs_AP_15 ├── Localizers_1 ├── MoCoSeries_19 ├── MoCoSeries_31 ├── Post_TMS_restingstate_30 ├── T1_mprage_1mm_13 ├── field_mapping_20 ├── field_mapping_21 └── restingstate_18Pull the HeuDiConv Docker container to your machine:
docker pull nipy/heudiconv
From a BASH shell (no, zsh will not do), navigate to the MRIS directory and run the
hdc_run.shscript for subject 219, session itbs, like this:./hdc_run.sh heuristic1.py 219 itbs
This should complete the conversion. After running, the Nifti directory will contain a bids-compliant subject directory:
└── sub-219 └── ses-itbs ├── anat ├── dwi ├── fmap └── funcThe following required BIDS text files are also created in the Nifti directory. Details for filling in these skeleton text files can be found under tabular files in the BIDS specification:
CHANGES README dataset_description.json participants.json participants.tsv task-rest_bold.json
Next, visit the bids validator.
Click Choose File and then select the Nifti directory. There should be no errors (though there are a couple of warnings).
Note
Your files are not uploaded to the BIDS validator, so there are no privacy concerns!
Look at the directory structure and files that were generated.
When you are ready, remove everything that was just created:
rm -rf Nifti/sub-* Nifti/.heudiconv Nifti/code/__pycache__ Nifti/*.json Nifti/*.tsv Nifti/README Nifti/CHANGE
Now you know what the results should look like.
In the following sections, you will build heuristic.py yourself so you can test different options and understand how to work with your own data.
Running HeuDiConv is a 3-step process
Step1 By passing some path information and flags to HeuDiConv, you generate a heuristic (translation) file skeleton and some associated descriptor text files. These all get placed in a hidden directory, .heudiconv under the Nifti directory.
Step2 Copy MRIS/Nifti/.heudiconv/heuristic.py to MRIS/Nifti/code/heuristic.py. You will modify the copied heuristic.py to specify BIDS output names and directories, and the input DICOM characteristics. Available input DICOM characteristics are listed in MRIS/Nifti/.heudiconv/dicominfo.tsv.
Step3 Having revised MRIS/Nifti/code/heuristic.py, you can now call HeuDiConv to run on more subjects and sessions. Each time you run it, additional subdirectories are created under .heudiconv that record the details of each subject (and session) conversion. Detailed provenance information is retained in the .heudiconv hidden directory. You can rename your heuristic file, which may be useful if you have multiple heuristic files for the same dataset.
TIPS
Name Directories as you wish: You can name the project directory (e.g., MRIS) and the output directory (e.g., Nifti) as you wish (just don’t put spaces in the names!).
IMA vs dcm: You are likely to have
IMAfiles if you copied your images from the scanner to an external drive, but.dcmfiles if you exported your images from Horos or Osirix. Watch out for capitalization differences in the sequence names (.dcmfiles are typically lower case, butIMAfiles are typically upper case).Age and Sex Extraction: Heudiconv will extract age and sex info from the DICOM header. If there is any reason to believe this information is wrong in the DICOM header (for example, it was made-up because no one knew how old the subject was, or it was considered a privacy concern), then you need to check the output. If you have Horos (or another DICOM editor), you can edit the values in the DICOM headers, otherwise you need to edit the values in the BIDS text file participants.tsv.
Separating Sessions: If you have multiple sessions at the scanner, you should create an Exam folder for each session. This will help you to keep the data organized and Exam will be reported in the study_description in your dicominfo.tsv, so that you can use it as a criterion.
Don’t manually combine DICOMS from different sessions: If you combine multiple sessions in one subject DICOM folder, heudiconv will fail to run and will complain about
conflicting study identifiers. You can get around the problem by figuring out which DICOMs are from different sessions and separating them so you deal with one set at a time. This may mean you have to manually edit the BIDS output.Why might you manually combine sessions you ask? Because you never intended to have multiple sessions, but the subject had to complete some scans the next day. Or, because the scanner had to be rebooted.
Don’t assume all your subjects’ dicoms have the same names or that the sequences were always run in the same order: If you develop a heuristic.py on one subject, try it and carefully evaluate the results on your other subjects. This is especially true if you already collected the data before you started thinking about automating the output. Every time you run HeuDiConv with heuristic.py, a new dicominfo.tsv file is generated. Inspect this for differences in protocol names and series descriptions etc.
Decompressing DICOMS: On 03/11/2019 I found that heudiconv failed if the data I exported from Horos was not decompressed. This was especially confusing because dcm2niix succeeded on this data…hmm.
Create unique DICOM protocol names at the scanner If you have the opportunity to influence the DICOM naming strategies, then try to ensure that there is a unique protocol name for every run. For example, if you repeat the fmri protocol three times, name the first one fmri_1, the next fmri_2, and the last fmri_3 (or any variation on this theme). This will make it much easier to uniquely specify the sequences when you convert and reduce your chance of errors.
Lesson 2: Step 1
From the MRIS directory, run the following Docker command to process the dcm files that you downloaded and unzipped for this tutorial. The subject number is 219:
docker run --rm -it -v ${PWD}:/base nipy/heudiconv:latest -d /base/dicom/{subject}/*/*/*.dcm -o /base/Nifti/ -f convertall -s 219 -c none
Warning
The above Docker command works in bash, but may not work in other shells, (e.g., zsh)
--rmmeans Docker should cleanup after itself-itmeans Docker should run interactively-v ${PWD}:/basebinds your current directory to/baseinside the container. You could also provide an absolute path to the MRIS directory.nipy/heudiconv:latestidentifies the Docker container to run (the latest version of heudiconv).-d /base/dicom/{subject}/*/*/*.dcmidentifies the path to the DICOM files and specifies that they have the extension.dcmin this case.-o /base/Nifti/is the output in Nifti. If the output directory does not exist, it will be created.-f convertallThis creates a heuristic.py template from an existing heuristic module. There are other heuristic modules , e.g., banda-bids.py, bids_with_ses.py, cmrr_heuristic.py, example.py, multires_7Tbold.py, reproin.py, studyforrest_phase2.py, test_reproin.py, uc_bids.py. heuristic.py is a good default though.-s 219specifies the subject number.219will replace {subject} in the-dargument when Docker actually runs.-c noneindicates you are not actually doing any conversion right now.Heudiconv generates a hidden directory MRIS/Nifti/.heudiconv/219/info and populates it with two files of interest: a skeleton heuristic.py and a dicominfo.tsv file.
After Step 1, the heuristic.py template contains explanatory text for you to read. I have removed this from heuristic1.py to keep it short.
Lesson 2: Step 2
You will modify three sections in heuristic.py. It is okay to rename this file, or to have several versions with different names. You just don’t want to mix up your new heuristic.py and the finished heuristic1.py while you are learning.
Your goal is to produce a working heuristic.py that will arrange the output in a BIDS directory structure. Once you create a working heuristic.py, you can run it for different subjects and sessions (keep reading).
I provide three section labels (1, 1b and 2) to facilitate exposition here. Each of these sections should be manually modified by you for your project.
Section 1
This heuristic.py does not import all sequences in the example Dicom directory. This is a feature of heudiconv: You do not need to import scouts, motion corrected images or other DICOMs of no interest.
You may wish to add, modify or remove keys from this section for your own data:
# Section 1: These key definitions should be revised by the user ################################################################### # For each sequence, define a key variables (e.g., t1w, dwi etc) and template using the create_key function: # key = create_key(output_directory_path_and_name). ###### TIPS ####### # If there are sessions, then session must be subfolder name. # Do not prepend the ses key to the session! It will be prepended automatically for the subfolder and the filename. # The final value in the filename should be the modality. It does not have a key, just a value. # Otherwise, there is a key for every value. # Filenames always start with subject, optionally followed by session, and end with modality. ###### Definitions ####### # The "data" key creates sequential numbers which can be used for naming sequences. # This is especially valuable if you run the same sequence multiple times at the scanner. data = create_key('run-{item:03d}') t1w = create_key('sub-{subject}/{session}/anat/sub-{subject}_{session}_T1w') dwi = create_key('sub-{subject}/{session}/dwi/sub-{subject}_{session}_dir-AP_dwi') # Save the RPE (reverse phase-encode) B0 image as a fieldmap (fmap). It will be used to correct # the distortion in the DWI fmap_rev_phase = create_key('sub-{subject}/{session}/fmap/sub-{subject}_{session}_dir-PA_epi') fmap_mag = create_key('sub-{subject}/{session}/fmap/sub-{subject}_{session}_magnitude') fmap_phase = create_key('sub-{subject}/{session}/fmap/sub-{subject}_{session}_phasediff') # Even if this is resting state, you still need a task key func_rest = create_key('sub-{subject}/{session}/func/sub-{subject}_{session}_task-rest_run-01_bold') func_rest_post = create_key('sub-{subject}/{session}/func/sub-{subject}_{session}_task-rest_run-02_bold')
Key
Define a short informative key variable name for each image sequence you wish to export. Note that you can use any key names you want (e.g. foo would work as well as fmap_phase), but you need to be consistent.
The
keyname is to the left of the=for each row in the above example.
Template
Use the variable
{subject}to make the code general purpose, so you can apply it to different subjects in Step 3.Use the variable
{session}to make the code general purpose only if you have multiple sessions for each subject.Once you use the variable
{session}:Ensure that a session gets added to the output path, e.g.,
sub-{subject}/{session}/anat/ANDSession gets added to the output filename:
sub-{subject}_{session}_T1wfor every image in the session.Otherwise you will get bids-validator errors.
Define the output directories and file names according to the BIDS specification
Note the output names for the fieldmap images (e.g., sub-219_ses-itbs_dir-PA_epi.nii.gz, sub-219_ses-itbs_magnitude1.nii.gz, sub-219_ses-itbs_magnitude2.nii.gz, sub-219_ses-itbs_phasediff.nii.gz).
The reverse_phase encode dwi image (e.g., sub-219_ses-itbs_dir-PA_epi.nii.gz) is grouped with the fieldmaps because it is used to correct other images.
Data that is not yet defined in the BIDS specification will cause the bids-validator to produce an error unless you include it in a .bidsignore file.
data
a key definition that creates sequential numbering
03dmeans create three slots for digits3d, and pad with zeros0.This is useful if you have a scanner sequence with a single name but you run it repeatedly and need to generate separate files for each run. For example, you might define a single functional sequence at the scanner and then run it several times instead of creating separate names for each run.
Note
It is usually better to name your sequences explicitly (e.g., run-01, run-02 etc.) rather than depending on sequential numbering. There will be less confusion later.
If you have a sequence with the same name that you run repeatedly WITHOUT the sequential numbering, HeuDiConv will overwrite earlier sequences with later ones.
To ensure that a sequence includes sequential numbering, you also need to add
run-{item:03d}(for example) to the key-value specification for that sequence.Here I illustrate with the t1w key-value pair:
If you started with:
t1w = create_key('sub-{subject}/anat/sub-{subject}_T1w'),
You could add sequence numbering like this:
t1w = create_key('sub-{subject}/anat/sub-{subject}_run-{item:03d}_T1w').
Now if you export several T1w images for the same subject and session, using the exact same protocol, each will get a separate run number like this:
sub-219_ses_run-001_T1w.nii.gz, sub-219_ses_run-002_T1w.nii.gz etc.
Section 1b
Based on your chosen keys, create a data dictionary called info:
# Section 1b: This data dictionary (below) should be revised by the user. ########################################################################### # info is a Python dictionary containing the following keys from the infotodict defined above. # This list should contain all and only the sequences you want to export from the dicom directory. info = {t1w: [], dwi: [], fmap_rev_phase: [], fmap_mag: [], fmap_phase: [], func_rest: [], func_rest_post: []} # The following line does no harm, but it is not part of the dictionary. last_run = len(seqinfo)
Enter each key in the dictionary in this format
key: [], for example,t1w: [].Separate the entries with commas as illustrated above.
Section 2
Define the criteria for identifying each DICOM series that corresponds to one of the keys you want to export:
# Section 2: These criteria should be revised by the user. ########################################################## # Define test criteria to check that each DICOM sequence is correct # seqinfo (s) refers to information in dicominfo.tsv. Consult that file for # available criteria. # Each sequence to export must have been defined in Section 1 and included in Section 1b. # The following illustrates the use of multiple criteria: for idx, s in enumerate(seqinfo): # Dimension 3 must equal 176 and the string 'mprage' must appear somewhere in the protocol_name if (s.dim3 == 176) and ('mprage' in s.protocol_name): info[t1w].append(s.series_id) # Dimension 3 must equal 74 and dimension 4 must equal 32, and the string 'DTI' must appear somewhere in the protocol_name if (s.dim3 == 74) and (s.dim4 == 32) and ('DTI' in s.protocol_name): info[dwi].append(s.series_id) # The string 'verify_P-A' must appear somewhere in the protocol_name if ('verify_P-A' in s.protocol_name): info[fmap_rev_phase] = [s.series_id] # Dimension 3 must equal 64, and the string 'field_mapping' must appear somewhere in the protocol_name if (s.dim3 == 64) and ('field_mapping' in s.protocol_name): info[fmap_mag] = [s.series_id] # Dimension 3 must equal 32, and the string 'field_mapping' must appear somewhere in the protocol_name if (s.dim3 == 32) and ('field_mapping' in s.protocol_name): info[fmap_phase] = [s.series_id] # The string 'resting_state' must appear somewhere in the protocol_name and the Boolean field is_motion_corrected must be False (i.e. not motion corrected) # This ensures I do NOT get the motion corrected MOCO series instead of the raw series! if ('restingstate' == s.protocol_name) and (not s.is_motion_corrected): info[func_rest].append(s.series_id) # The string 'Post_TMS_resting_state' must appear somewhere in the protocol_name and the Boolean field is_motion_corrected must be False (i.e. not motion corrected) # This ensures I do NOT get the motion corrected MOCO series instead of the raw series. if ('Post_TMS_restingstate' == s.protocol_name) and (not s.is_motion_corrected): info[func_rest_post].append(s.series_id)
To define the criteria, look at dicominfo.tsv in .heudiconv/info. This file contains tab-separated values so you can easily view it in Excel or any similar spreadsheet program. dicominfo.tsv is not used programmatically to run heudiconv (i.e., you could delete it with no adverse consequences), but it is very useful for defining the test criteria for Section 2 of heuristic.py.
Some values in dicominfo.tsv might be wrong. For example, my reverse phase encode sequence with two acquisitions of 74 slices each is reported as one acquisition with 148 slices (2018_12_11). Hopefully they’ll fix this. Despite the error in dicominfo.tsv, dcm2niix reconstructed the images correctly.
You will be adding, removing or altering values in conditional statements based on the information you find in dicominfo.tsv.
seqinfo(s) refers to the same information you can view in dicominfo.tsv (although seqinfo does not rely on dicominfo.tsv).Here are two types of criteria:
s.dim3 == 176is an equivalence (e.g., good for checking dimensions for a numerical data type). For our sample T1w image to be exported from DICOM, it must have 176 slices in the third dimension.'mprage' in s.protocol_namesays the protocol name string must include the word mprage for the T1w image to be exported from DICOM. This criterion string is case-sensitive.
info[t1w].append(s.series_id)Given that the criteria are satisfied, the series should be named and organized as described in Section 1 and referenced by the info dictionary. The information about the processing steps is saved in the .heudiconv subdirectory.Here I have organized each conditional statement so that the sequence protocol name comes first followed by other criteria if relevant. This is not necessary, though it does make the resulting code easier to read.
Lesson 2: Step 3
You have now done all the hard work for your project. When you want to add a subject or session, you only need to run this third step for that subject or session (A record of each run is kept in .heudiconv for you):
docker run --rm -it -v ${PWD}:/base nipy/heudiconv:latest -d /base/dicom/{subject}/*/*.dcm -o /base/Nifti/ -f /base/Nifti/code/heuristic.py -s 219 -ss itbs -c dcm2niix -b --minmeta --overwrite
Warning
The above Docker command WORKS IN BASH, but may not work in other shells! For example, zsh is upset by the form {subject} but bash actually doesn’t mind.
The first time you run this step, several important text files are generated (e.g., CHANGES, dataset_description.json, participants.tsv, README etc.). On subsequent runs, information may be added (e.g., participants.tsv will be updated). Other files, like the README and dataset_description.json should be filled in manually after they are first generated.
This Docker command is slightly different from the previous Docker command you ran.
-f /base/Nifti/code/heuristic.pynow tells HeuDiConv to use your revised heuristic.py in the code directory.In this case, we specify the subject we wish to process
-s 219and the name of the session-ss itbs.We could specify multiple subjects like this:
-s 219 220 -ss itbs
-c dcm2niix -bindicates that we want to use the dcm2niix converter with the -b flag (which creates BIDS).--minmetaensures that only the minimum necessary amount of data gets added to the JSON file when created. On the off chance that there is a LOT of meta-information in the DICOM header, the JSON file will not get swamped by it. fmriprep and mriqc are very sensitive to this information overload and will crash, so minmeta provides a layer of protection against such corruption.--overwriteThis is a peculiar option. Without it, I have found the second run of a sequence does not get generated. But with it, everything gets written again (even if it already exists). I don’t know if this is my problem or the tool…but for now, I’m using--overwrite.Step 3 should produce a tree like this:
Nifti ├── CHANGES ├── README ├── code │ ├── __pycache__ │ │ └── heuristic1.cpython-36.pyc │ ├── heuristic1.py │ └── heuristic2.py ├── dataset_description.json ├── participants.json ├── participants.tsv ├── sub-219 │ └── ses-itbs │ ├── anat │ │ ├── sub-219_ses-itbs_T1w.json │ │ └── sub-219_ses-itbs_T1w.nii.gz │ ├── dwi │ │ ├── sub-219_ses-itbs_dir-AP_dwi.bval │ │ ├── sub-219_ses-itbs_dir-AP_dwi.bvec │ │ ├── sub-219_ses-itbs_dir-AP_dwi.json │ │ └── sub-219_ses-itbs_dir-AP_dwi.nii.gz │ ├── fmap │ │ ├── sub-219_ses-itbs_dir-PA_epi.json │ │ ├── sub-219_ses-itbs_dir-PA_epi.nii.gz │ │ ├── sub-219_ses-itbs_magnitude1.json │ │ ├── sub-219_ses-itbs_magnitude1.nii.gz │ │ ├── sub-219_ses-itbs_magnitude2.json │ │ ├── sub-219_ses-itbs_magnitude2.nii.gz │ │ ├── sub-219_ses-itbs_phasediff.json │ │ └── sub-219_ses-itbs_phasediff.nii.gz │ ├── func │ │ ├── sub-219_ses-itbs_task-rest_run-01_bold.json │ │ ├── sub-219_ses-itbs_task-rest_run-01_bold.nii.gz │ │ ├── sub-219_ses-itbs_task-rest_run-01_events.tsv │ │ ├── sub-219_ses-itbs_task-rest_run-02_bold.json │ │ ├── sub-219_ses-itbs_task-rest_run-02_bold.nii.gz │ │ └── sub-219_ses-itbs_task-rest_run-02_events.tsv │ ├── sub-219_ses-itbs_scans.json │ └── sub-219_ses-itbs_scans.tsv └── task-rest_bold.json
Exploring Criteria
dicominfo.tsv contains a human readable version of seqinfo. Each column of data can be used as criteria for identifying the correct DICOM image. We have already provided examples of using string types, numbers, and Booleans (True-False). Tuples (immutable lists) are also available and examples of using these are provided below. To ensure that you are extracting the images you want, you need to be very careful about creating your initial heuristic.py.
Why Experiment?
Criteria can be tricky. Ensure the NIfTI files you create are the correct ones (for example, not the derived or motion corrected if you didn’t want that). In addition to looking at the images created (which tells you whether you have a fieldmap or T1w etc.), you should look at the dimensions of the image. Not only the dimensions, but the range of intensity values and the size of the image on disk should match for dcm2niix and heudiconv’s heuristic.py.
For really tricky cases, download and install dcm2niix on your local machine and run it for a sequence of concern (in my experience, it is usually fieldmaps that go wrong).
Although Python does not require you to use parentheses while defining criteria, parentheses are a good idea. Parentheses will help ensure that complex criteria involving multiple logical operators
and, or, notmake sense and behave as expected.
Tuples
Suppose you want to use the values in the field image_type? It is not a number or string or Boolean. To discover the data type of a column, you can add a statement like this print(type(s.image_type)) to the for loop in Section 2 of heuristic.py. Then run heuristic.py (preferably without any actual conversions) and you should see an output like this <class 'tuple'>. Here is an example of using a value from image_type as a criterion:
if ('ASL_3D_tra_iso' == s.protocol_name) and ('TTEST' in s.image_type):
info[asl_der].append(s.series_id)
Note that this differs from testing for a string because you cannot test for any substring (e.g., ‘TEST’ would not work). String tests will not work on a tuple datatype.
Note
image_type is described in the DICOM specification
Lesson 3: reproin.py
If you don’t want to modify a Python file as you did for heuristic.py, an alternative is to name your image sequences at the scanner using the reproin naming convention. Take some time getting the scanner protocol right, because it is the critical job for running reproin. Then a single Docker command converts your DICOMS to the BIDS data structure. There are more details about reproin in the Links section above.
You should already have Docker installed and have downloaded HeuDiConv as described in Lesson 1.
Download and unzip the phantom dataset: reproin_dicom.zip generated here at the University of Arizona on our Siemens Skyra 3T with Syngo MR VE11c software on 2018_02_08.
You should see a new directory REPROIN. This is a simple reproin-compliant dataset without sessions. Derived dwi images (ADC, FA etc.) that the scanner produced were removed.
Change directory to REPROIN. The directory structure should look like this:
REPROIN ├── data └── dicom └── 001 └── Patterson_Coben\ -\ 1 ├── Localizers_4 ├── anatT1w_acqMPRAGE_6 ├── dwi_dirAP_9 ├── fmap_acq4mm_7 ├── fmap_acq4mm_8 ├── fmap_dirPA_15 └── func_taskrest_16From the REPROIN directory, run this Docker command:
docker run --rm -it -v ${PWD}:/base nipy/heudiconv:latest -f reproin --bids -o /base/data --files /base/dicom/001 --minmeta--rmmeans Docker should cleanup after itself-itmeans Docker should run interactively-v ${PWD}:/basebinds your current directory to/baseinside the container. Alternatively, you could provide an absolute path to the REPROIN directory.nipy/heudiconv:latestidentifies the Docker container to run (the latest version of heudiconv).-f reproinspecifies the converter file to use-o /base/data/specifies the output directory data. If the output directory does not exist, it will be created.--files /base/dicom/001identifies the path to the DICOM files.--minmetaensures that only the minimum necessary amount of data gets added to the JSON file when created. On the off chance that there is a LOT of meta-information in the DICOM header, the JSON file will not get swamped by it. fmriprep and mriqc are very sensitive to this information overload and will crash, so minmeta provides a layer of protection against such corruption.
That’s it. Below we’ll unpack what happened.
Output Directory Structure
Reproin produces a hierarchy of BIDS directories like this:
data
└── Patterson
└── Coben
├── sourcedata
│ └── sub-001
│ ├── anat
│ ├── dwi
│ ├── fmap
│ └── func
└── sub-001
├── anat
├── dwi
├── fmap
└── func
The dataset is nested under two levels in the output directory: Region (Patterson) and Exam (Coben). Tree is reserved for other purposes at the UA research scanner.
Although the Program Patient is not visible in the output hierarchy, it is important. If you have separate sessions, then each session should have its own Program name.
sourcedata contains tarred gzipped (tgz) sets of DICOM images corresponding to each NIFTI image.
sub-001 contains the BIDS dataset.
The hidden directory is generated: REPROIN/data/Patterson/Coben/.heudiconv.
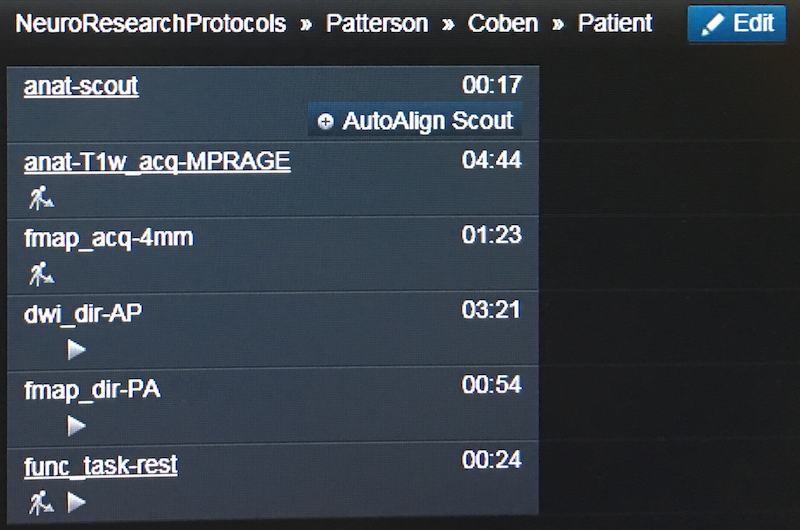
At the Scanner
Here is this phantom dataset displayed in the scanner dot cockpit. The directory structure is defined at the top: Patterson >> Coben >> Patient
Region = Patterson
Exam = Coben
Program = Patient
Reproin Scanner File Names
For both BIDS and reproin, names are composed of an ordered series of key-value pairs. Each key and its value are joined with a dash
-(e.g.,acq-MPRAGE,dir-AP). These key-value pairs are joined to other key-value pairs with underscores_. The exception is the modality label, which is discussed more below.Reproin scanner sequence names are simplified relative to the final BIDS output and generally conform to this scheme (but consult the reference for additional options):
sequence type-modality label_session-session name_task-task name_acquisition-acquisition detail_run-run number_direction-direction label:| func-bold_ses-pre_task-faces_acq-1mm_run-01_dir-AP
Each sequence name begins with the seqtype key. The seqtype key is the modality and corresponds to the name of the BIDS directory where the sequence belongs, e.g.,
anat,dwi,fmaporfunc.The seqtype key is optionally followed by a dash
-and a modality label value (e.g.,anat-scoutoranat-T2W). Often, the modality label is not needed because there is a predictable default for most seqtypes:For anat the default modality is
T1W. Thus a sequence namedanatwill have the same output BIDS files as a sequence namedanat-T1w: sub-001_T1w.nii.gz.For fmap the default modality is
epi. Thusfmap_dir-PAwill have the same output asfmap-epi_dir-PA: sub-001_dir-PA_epi.nii.gz.For func the default modality is
bold. Thus,func-bold_task-restwill have the same output asfunc_task-rest: sub-001_task-rest_bold.nii.gz.Reproin gets the subject number from the DICOM metadata.
If you have multiple sessions, the session name does not need to be included in every sequence name in the program (i.e., Program= Patient level mentioned above). Instead, the session can be added to a single sequence name, usually the scout (localizer) sequence e.g.
anat-scout_ses-pre, and reproin will propagate the session information to the other sequence names in the Program. Interestingly, reproin does not add the localizer to your BIDS output.When our scanner exports the DICOM sequences, all dashes are removed. But don’t worry, reproin handles this just fine.
In the UA phantom reproin data, the subject was named
01. Horos reports the subject number as01but exports the DICOMS into a directory001. If the data are copied to an external drive at the scanner, then the subject number is reported as001_001and the images are*.IMAinstead of*.dcm. Reproin does not care, it handles all of this gracefully. Your output tree (excluding sourcedata and .heudiconv) should look like this:. |-- CHANGES |-- README |-- dataset_description.json |-- participants.tsv |-- sub-001 | |-- anat | | |-- sub-001_acq-MPRAGE_T1w.json | | `-- sub-001_acq-MPRAGE_T1w.nii.gz | |-- dwi | | |-- sub-001_dir-AP_dwi.bval | | |-- sub-001_dir-AP_dwi.bvec | | |-- sub-001_dir-AP_dwi.json | | `-- sub-001_dir-AP_dwi.nii.gz | |-- fmap | | |-- sub-001_acq-4mm_magnitude1.json | | |-- sub-001_acq-4mm_magnitude1.nii.gz | | |-- sub-001_acq-4mm_magnitude2.json | | |-- sub-001_acq-4mm_magnitude2.nii.gz | | |-- sub-001_acq-4mm_phasediff.json | | |-- sub-001_acq-4mm_phasediff.nii.gz | | |-- sub-001_dir-PA_epi.json | | `-- sub-001_dir-PA_epi.nii.gz | |-- func | | |-- sub-001_task-rest_bold.json | | |-- sub-001_task-rest_bold.nii.gz | | `-- sub-001_task-rest_events.tsv | `-- sub-001_scans.tsv `-- task-rest_bold.json
Note that despite all the the different subject names (e.g.,
01,001and001_001), the subject is labeledsub-001.